
Protein located in the wrong part of the cell can contribute to several diseases such as Alzheimer’s disease, cystic fibrosis and cancer. In one human cell, however, there are about 70,000 different proteins and protein variants, and since scientists can usually test only a handful in one experience, it is very expensive and time -consuming manual protein identification.
The new generation of computing techniques is trying to streamline the process using machine learning models that often use data sets containing thousands of proteins and their location, measured on multiple cell lines. One of the large such data sets is the atlas of human protein, which catalogizes subcellular behavior of more than 13,000 proteins in more than 40 cell lines. But as huge as it is, the human protein atlas has only explored about 0.25 percent of all possible pairs of all proteins and cell lines in the database.
Now scientists from MIT, Harvard University and Broad Institute of Mit and Harvard have developed a new computing approach that can effectively explore the remaining uncharted space. Their method can predict the rent of any protein in any human cell line, although protein and cells have never been tested.
Their technique is a step further than many methods based on AI localization of protein levels at the single -cell level, rather than an average estimated estimate across all cell -type cells. For example, this unicellular localization could, for example, to determine the lease of protein to a specific cancer cell.
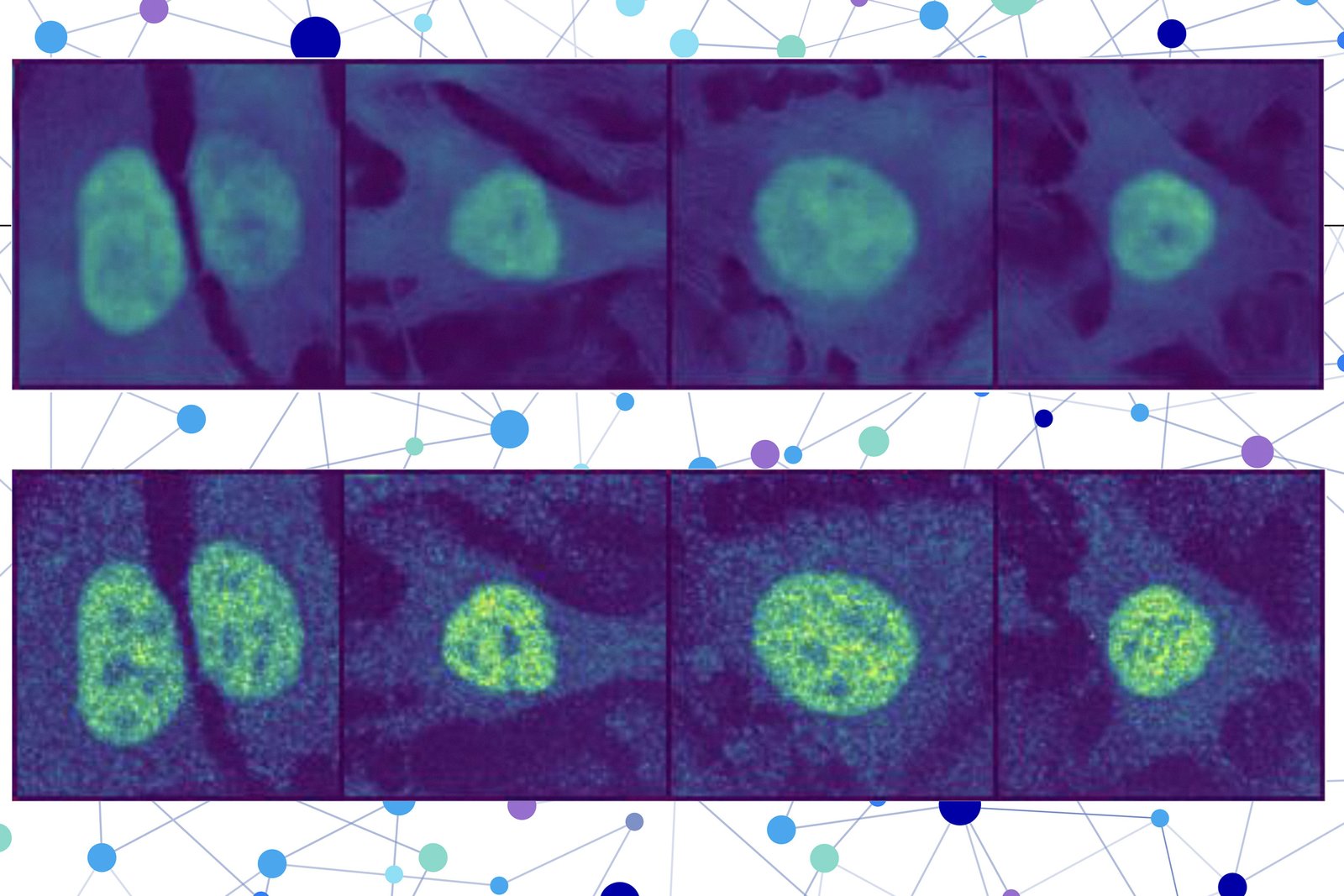
Scientists combined a model of protein language with a special type of computer vision model that captured rich details of protein and cell. Finally, the user receives a cell image with a highlighted part indicating the prediction of the model where the protein is located. The sale of protein localization testifies to its functional condition, this technique could help scientists and clinical doctors to diagnose or identify the objectives of the drug more effectively, while allowing biologists to understand how complex biological processes are related to the location of proteins.
“You could perform this computer’s protein experiment without having to touch any laboratory benches, hopefully you will save the months of effort. Even if you still have to verify prediction, this technique could act as initial screening of what to test at Yitong TSEO experience, postgraduate student holding this research. ”
TSEO joined the article by the author Xinyi Zhang, a postgraduate student at the Department of Electrical Engineering and Informatics (EECS) and Eric and Wendy Schmidt Center at the Broad Institute; Yunhao Bai Broad Institute; and the head of the Fei Chen, Associate Professor at Harvard and a member of the Wide Institute and Caroline Uhler, Andrew and Erna Viterbi Professor ECS and MIT Institute for Data, Systems and Society), which is also Eric and Wendy Schmidt and Mit’s Laboratory (Humans). Research will appear today Nature methods.
The cooperation of models
Many existing protein prediction models can only create predictions based on protein and cell data on which they were trained or unusable for determining the persecution of protein in one cell.
To overcome these restrictions, scientists have created a two -piece method for the prediction of subcellular lease of invisible proteins called Pips.
The first part uses the model of the protein sequence to capture the properties determining the location of the protein and its 3D structure based on the acid chain.
The second part includes the model insertion of the image that is designed to fill in the missing part of the image. This model of computer vision focuses on three colored cell images to collect information about the condition of this cell, such as its type, individual features and where it is under stress.
PUPS connects to the reprints created by each model to predict where the protein is placed in one cell, using the image decoder to perform a highlighted image that shows the expected rental.
“Different cells in the cell line show characteristics and our model is able to understand this nuances,” says Tseo.
The user enters an amino acid sequence that consists of proteins and three cellular spots – one for the core, one for microtubules and one for endoplasmic reticulum. Then puppies des the rest.
Desperate understanding
During the training process, scientists used several tricks to teach pop to combine information from each model in a way that an educated estimate of the persecution of protein can, even if this protein has been seen earlier.
For example, they allocate the model secondary task during training: explicitly name the localization section, such as a cell nucleus. This is therefore, along with the primary task of the entry to help the model more effectively to learn.
A good analogy can be a teacher who asks their tips to draw all parts of the flower in addition to writing their names. It has been found that this next step helps the model to improve its general understanding of possible cellular compartments.
In addition, the fact that PUB is trained on proteins and cell lines at the same time helps to develop de -buy understanding where they tend to locate in the proteins of the cellular image.
Puppies can even understand the ont, how different parts of Protein’s sequences contribute to separals to its overall location.
“Most other methods usually require you to first have a protein stain, so you have already seen in your training sessions. Our approval is unique in that it can generalize across proteins and cell lines at the same time,” Zhang says.
Becuse PUPS can generalize to invisible proteins, it can capture a change in localization powered by unique protein mutations that are not included in the atlas of human protein.
Scientists have verified that puppies could predict subcellular rent of new proteins in invisible cell lines by performing laboratory experiments and comparing the results. In addition, in comparison with the basic AI method, pubs showed an average of a minor prediction error across the proteins they tested.
In the future, scientists want to strengthen puppies so that the model can understand the interactions of protein-protein and create predictions of localization for multiple proteins in the cell. In the longer term, puppies want to allow predictions in terms of living human tissue rather than refined cells.
This research is financed by the Eric and Wendy Schmidt center at the Broad Institute, the National Health Constitution, the National Science Foundation, Burroughs Welcome Fund, the Searle Scholars Foundation, the Harvard STEM Cell Institute, the Maritime Research Office and the Ministry of Energy.